Strelka 2Pass analysis (CRAM)¶
Strelka2PassWorkflowCram · 1 contributor · 1 version
- This is the full 2pass analysis workflow to do joint somatic variant calling with strelka2.
The idea is similar to the RNASeq 2pass analysis, when the input of the first analysis is used to guide the second analysis.
- The workflow will
- run manta
- run strelka with manata output
- run strelka with strelka and manta output
- reannotate the filter column
- output resuults
Quickstart¶
from janis_bioinformatics.tools.dawson.workflows.variantcalling.multisample.strelka2.strelka2passworkflow_cram import Strelka2PassWorkflowCram wf = WorkflowBuilder("myworkflow") wf.step( "strelka2passworkflowcram_step", Strelka2PassWorkflowCram( normalBam=None, tumorBams=None, reference=None, ) ) wf.output("snvs", source=strelka2passworkflowcram_step.snvs) wf.output("indels", source=strelka2passworkflowcram_step.indels) wf.output("svs", source=strelka2passworkflowcram_step.svs)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for Strelka2PassWorkflowCram:
# user inputs
janis inputs Strelka2PassWorkflowCram > inputs.yaml
inputs.yaml
normalBam: normalBam.cram
reference: reference.fasta
tumorBams:
- tumorBams_0.cram
- tumorBams_1.cram
- Run Strelka2PassWorkflowCram with:
janis run [...run options] \
--inputs inputs.yaml \
Strelka2PassWorkflowCram
Information¶
URL: No URL to the documentation was provided
| ID: | Strelka2PassWorkflowCram |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | 0.2 |
| Authors: | Sebastian Hollizeck |
| Citations: | |
| Created: | 2019-10-11 |
| Updated: | 2020-12-10 |
Outputs¶
| name | type | documentation |
|---|---|---|
| snvs | Array<Gzipped<VCF>> | |
| indels | Array<Gzipped<VCF>> | |
| svs | Array<Optional<Gzipped<VCF>>> |
Workflow¶
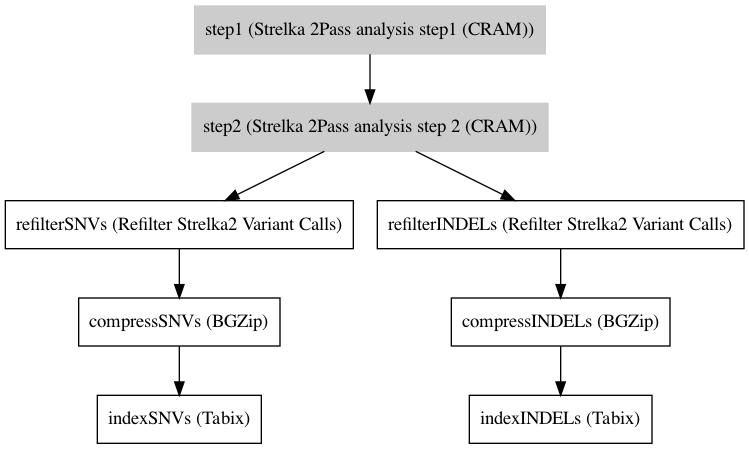
Embedded Tools¶
| Strelka 2Pass analysis step1 (CRAM) | Strelka2PassWorkflowStep1Cram/0.1.1 |
| Strelka 2Pass analysis step 2 (CRAM) | Strelka2PassWorkflowStep2Cram/0.1.1 |
| Refilter Strelka2 Variant Calls | refilterStrelka2Calls/0.1.8 |
| BGZip | bgzip/1.2.1 |
| Tabix | tabix/1.2.1 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| normalBam | CramPair | The bam of the normal sample. Strelka will assign any read in this bam to the normal sample, even if this bam contains multiple samples |
| tumorBams | Array<CramPair> | The bam of the tumour sample. Strelka will assign any read in this bam to the normal sample, even if this bam contains multiple samples |
| reference | FastaFai | The fai indexed fasta reference, the bams were aligned to. |
| configStrelka | Optional<File> | The possibly changed ini to use for Strelka2. This can be used to skip regions with extreme depth, like in heterochromatin regions, which lead to very long runtimes. |
| callRegions | Optional<Gzipped<bed>> | The tabix indexed bed file of regions to restict the analysis on. If this is unset, every site in the genome will be analysed. |
| exome | Optional<Boolean> | Sets the flag to analyse everything in exome mode. This will adjust the parameter for a non uniform coverage profile. |
| sampleNames | Optional<Array<String>> | The names of the tumour samples. This will only be used to rename output files. if unset, the output will be numbered in the same order as the input files. |
| minAD | Optional<Integer> | Minimum read support for a variant to be considered a true variant. |
Workflow Description Language¶
version development
import "tools/Strelka2PassWorkflowStep1Cram_0_1_1.wdl" as S
import "tools/Strelka2PassWorkflowStep2Cram_0_1_1.wdl" as S2
import "tools/refilterStrelka2Calls_0_1_8.wdl" as R
import "tools/bgzip_1_2_1.wdl" as B
import "tools/tabix_1_2_1.wdl" as T
workflow Strelka2PassWorkflowCram {
input {
File normalBam
File normalBam_crai
Array[File] tumorBams
Array[File] tumorBams_crai
File reference
File reference_fai
File? configStrelka
File? callRegions
File? callRegions_tbi
Boolean? exome = false
Array[String]? sampleNames
Int? minAD = 2
}
scatter (t in transpose([tumorBams, tumorBams_crai])) {
call S.Strelka2PassWorkflowStep1Cram as step1 {
input:
normalBam=normalBam,
normalBam_crai=normalBam_crai,
tumorBam=t[0],
tumorBam_crai=t[1],
reference=reference,
reference_fai=reference_fai,
callRegions=callRegions,
callRegions_tbi=callRegions_tbi,
exome=select_first([exome, false]),
configStrelka=configStrelka
}
}
scatter (t in transpose([tumorBams, tumorBams_crai])) {
call S2.Strelka2PassWorkflowStep2Cram as step2 {
input:
normalBam=normalBam,
normalBam_crai=normalBam_crai,
tumorBam=t[0],
tumorBam_crai=t[1],
reference=reference,
reference_fai=reference_fai,
callRegions=callRegions,
callRegions_tbi=callRegions_tbi,
exome=select_first([exome, false]),
configStrelka=configStrelka,
indelCandidates=step1.candIndels,
indelCandidates_tbi=step1.candIndels_tbi,
strelkaSNVs=step1.snvs,
strelkaSNVs_tbi=step1.snvs_tbi
}
}
call R.refilterStrelka2Calls as refilterSNVs {
input:
inputFiles=step2.snvs,
inputFiles_tbi=step2.snvs_tbi,
minAD=select_first([minAD, 2]),
sampleNames=sampleNames
}
scatter (r in refilterSNVs.out) {
call B.bgzip as compressSNVs {
input:
file=r
}
}
scatter (c in compressSNVs.out) {
call T.tabix as indexSNVs {
input:
inp=c
}
}
call R.refilterStrelka2Calls as refilterINDELs {
input:
inputFiles=step2.indels,
inputFiles_tbi=step2.indels_tbi,
minAD=select_first([minAD, 2]),
sampleNames=sampleNames
}
scatter (r in refilterINDELs.out) {
call B.bgzip as compressINDELs {
input:
file=r
}
}
scatter (c in compressINDELs.out) {
call T.tabix as indexINDELs {
input:
inp=c
}
}
output {
Array[File] snvs = indexSNVs.out
Array[File] snvs_tbi = indexSNVs.out_tbi
Array[File] indels = indexINDELs.out
Array[File] indels_tbi = indexINDELs.out_tbi
Array[File?] svs = step1.somaticSVs
Array[File?] svs_tbi = step1.somaticSVs_tbi
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Strelka 2Pass analysis (CRAM)
doc: |-
This is the full 2pass analysis workflow to do joint somatic variant calling with strelka2.
The idea is similar to the RNASeq 2pass analysis, when the input of the first analysis is used to guide the second analysis.
The workflow will
* run manta
* run strelka with manata output
* run strelka with strelka and manta output
* reannotate the filter column
* output resuults
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: ScatterFeatureRequirement
- class: SubworkflowFeatureRequirement
inputs:
- id: normalBam
doc: |-
The bam of the normal sample. Strelka will assign any read in this bam to the normal sample, even if this bam contains multiple samples
type: File
secondaryFiles:
- pattern: .crai
- id: tumorBams
doc: |-
The bam of the tumour sample. Strelka will assign any read in this bam to the normal sample, even if this bam contains multiple samples
type:
type: array
items: File
secondaryFiles:
- pattern: .crai
- id: reference
doc: The fai indexed fasta reference, the bams were aligned to.
type: File
secondaryFiles:
- pattern: .fai
- id: configStrelka
doc: |-
The possibly changed ini to use for Strelka2. This can be used to skip regions with extreme depth, like in heterochromatin regions, which lead to very long runtimes.
type:
- File
- 'null'
- id: callRegions
doc: |-
The tabix indexed bed file of regions to restict the analysis on. If this is unset, every site in the genome will be analysed.
type:
- File
- 'null'
secondaryFiles:
- pattern: .tbi
- id: exome
doc: |-
Sets the flag to analyse everything in exome mode. This will adjust the parameter for a non uniform coverage profile.
type: boolean
default: false
- id: sampleNames
doc: |-
The names of the tumour samples. This will only be used to rename output files. if unset, the output will be numbered in the same order as the input files.
type:
- type: array
items: string
- 'null'
- id: minAD
doc: Minimum read support for a variant to be considered a true variant.
type: int
default: 2
outputs:
- id: snvs
type:
type: array
items: File
outputSource: indexSNVs/out
- id: indels
type:
type: array
items: File
outputSource: indexINDELs/out
- id: svs
type:
type: array
items:
- File
- 'null'
outputSource: step1/somaticSVs
steps:
- id: step1
label: Strelka 2Pass analysis step1 (CRAM)
in:
- id: normalBam
source: normalBam
- id: tumorBam
source: tumorBams
- id: reference
source: reference
- id: callRegions
source: callRegions
- id: exome
source: exome
- id: configStrelka
source: configStrelka
scatter:
- tumorBam
run: tools/Strelka2PassWorkflowStep1Cram_0_1_1.cwl
out:
- id: diploid
- id: candIndels
- id: indels
- id: snvs
- id: somaticSVs
- id: step2
label: Strelka 2Pass analysis step 2 (CRAM)
in:
- id: normalBam
source: normalBam
- id: tumorBam
source: tumorBams
- id: reference
source: reference
- id: callRegions
source: callRegions
- id: exome
source: exome
- id: configStrelka
source: configStrelka
- id: indelCandidates
source: step1/candIndels
- id: strelkaSNVs
source: step1/snvs
scatter:
- tumorBam
run: tools/Strelka2PassWorkflowStep2Cram_0_1_1.cwl
out:
- id: indels
- id: snvs
- id: refilterSNVs
label: Refilter Strelka2 Variant Calls
in:
- id: inputFiles
source: step2/snvs
- id: minAD
source: minAD
- id: sampleNames
source: sampleNames
run: tools/refilterStrelka2Calls_0_1_8.cwl
out:
- id: out
- id: compressSNVs
label: BGZip
in:
- id: file
source: refilterSNVs/out
scatter:
- file
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: indexSNVs
label: Tabix
in:
- id: inp
source: compressSNVs/out
scatter:
- inp
run: tools/tabix_1_2_1.cwl
out:
- id: out
- id: refilterINDELs
label: Refilter Strelka2 Variant Calls
in:
- id: inputFiles
source: step2/indels
- id: minAD
source: minAD
- id: sampleNames
source: sampleNames
run: tools/refilterStrelka2Calls_0_1_8.cwl
out:
- id: out
- id: compressINDELs
label: BGZip
in:
- id: file
source: refilterINDELs/out
scatter:
- file
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: indexINDELs
label: Tabix
in:
- id: inp
source: compressINDELs/out
scatter:
- inp
run: tools/tabix_1_2_1.cwl
out:
- id: out
id: Strelka2PassWorkflowCram